"*" indicates required fields


Apert Syndrome Treatment in Dallas, TX
The International Craniofacial Institute in Dallas, Texas is recognized worldwide and has treated patients from over 30 countries around the globe. The ultimate goal is to provide patients with the best possible care in aesthetic and functional corrections. Each patient is treated as an individual with specific needs, and the treatment plan is always tailored according to their medical, surgical, dental, and psychosocial demands imposed by their circumstances. One of the conditions treated by our finest team of doctors is Apert syndrome which occurs with a rate of one in 160,000 live births.
What Is Apert Syndrome?
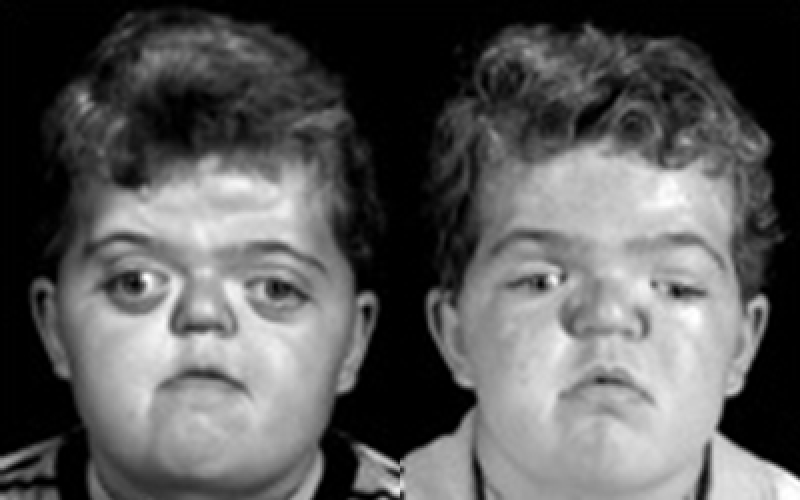
Apert syndrome is a genetic anomaly that occurs at the embryonic stage. Infants born with this condition have a distorted shape of skull and face. The bones of the skull fuse prematurely, causing the appearance of a sunken face, wide-set eyes, and beaked nose. In addition, upper or lower jaw remains underdeveloped, leading to crowding of the teeth and other dental issues. In many cases, the development of the brain is hindered, causing disruption with intellectual growth. Shallow and weak eye sockets frequently lead to problematic vision discrepancies. Skull malformations cannot be cured, but surgery can successfully correct some of the aesthetics. Other symptoms often accompany Apert syndrome such as webbed hands and feet along with gastrointestinal, urinary, and heart problems.
How Do People Inherit Apert Syndrome?
Apert syndrome is inherited in a pattern of altered genes. Almost all individuals affected with this syndrome exhibit signs of new gene mutations prevalent enough to cause the disorder. In most cases, people affected with Apert do not have a family history of the syndrome. However, the altered gene can be inherited by the next generation.
How Common Is Apert Syndrome?
Conflicting statistics show an array of different numbers. Recent studies show that Apert affects one infant in every 160,000 newborns. The disease is rare, and over 98 percent of cases surface because of new gene mutations.
Apert Syndrome Characteristics
Abnormal fusion of the bones causes several visible characteristics specific to the Apert syndrome, including the following:
- Unusual shape of the head
- Wide-set eyes with shallow sockets and poorly closing eyelids
- Recessed mid-face
- Beak-shaped nose
- Underdeveloped jaws with underbite and crowded dentition
- Cleft palate
- Impaired hearing capabilities
- Atypical spine development
- Fused fingers and toes
- Limited intellectual development
- Gastrointestinal malfunctions
- Cardiac malformations
- Hyperhydrosis (unusually heavy sweating)
- Severe acne
- Patches of lost hair
What Is the Cause of Apert Syndrome?
The main cause of developing Apert is a mutated gene that is responsible for guiding the bones to fuse beginning in the womb. A protein called fibroblast growth factor receptor 2 (FGFR 2) prompts immature cells to transform into bone cells. When the protein is altered during the embryonic process, a premature bone fusion occurs, leading to disfigurations characteristic of Apert syndrome. Abnormal growth patterns develop as the soft sutures of the skull close in an untimely manner. This type of gene alteration is sporadic and random. If neither of the parents carries the gene, the child will most likely develop without the symptoms of Apert. If one of the parents is affected, the newborn has a 50 percent chance of inheriting the syndrome as well.
Apert Syndrome Treatment
The treatment plan usually consists of several steps during different stages of maturity and may include partial re-arrangement of fused skull bones to release the pressure, palatal closure, and mid-face and eye surgeries. In addition, speech therapy and orthodontic intervention may be necessary. The child’s response to surgery is closely monitored to observe the progress and determine the need for any future surgical approach. Each change is carefully identified and promptly addressed as needed.
The goal of each surgery is to improve the child’s breathing, swallowing, and speaking patterns. It is also focused on the enhancement of brain function, central nervous system, and all senses. At every level of maturity, the child’s progress is evaluated, and it often opens a new opportunity for additional reconstruction.
Why Choose International Craniofacial Institute?
The International Craniofacial Institute in Dallas, Texas is a leading institute for the accurate diagnosis and quality treatment of Apert syndrome and other syndromes and conditions. Our institute was founded in 1971 by Dr. Kenneth Salyer, a surgeon. Today, the institute is organized and led by the director, Dr. David G. Genecov. Dr. Genecov operates the International Craniofacial Institute as one of the nation’s most prestigious centers for palate repair, craniofacial repair, and cleft lip repair.
At our institute, we train doctors and surgeons from all over the world. In addition, our doctors have treated more than 17000 patients. These patients come from the United States, as well as other countries.
To alter and correct craniofacial abnormalities and difficulties, a high skill set is demanded, and we have that here. Our doctors, surgeons and the rest of the staff are extremely knowledgeable and always up to date on the newest methods of diagnosis and treatment. Among all of our employees, we have decades of experience working with different syndromes, including Apert syndrome.
If you have a child or another family member who is suffering from a genetic syndrome or has a cleft lip, cleft palate, or craniofacial complication, the staff at the International Craniofacial Institute can help. Contact us today to talk with the doctors and staff about your options and how we can help.






